A cisztás fibrózis és szűrése
65 rózsa, így nevezik világszerte ezt a betegséget. Az elnevezés egy kanadai kórházból származik, ahol a cisztás fibrózissal kezelt kisgyerekek nem tudták kimondani a betegség angol nevét (cystic fibrosis), ezért helyette a sixty-five roses kifejezést használták, amely jelentése 65 rózsa. Európában ez a leggyakoribb öröklött genetikai betegség, minden 25. ember hordoz valamilyen cisztás fibrózis mutációt, ezért az ezzel kapcsolatos hordozóságszűrés a várandósság előtt, vagy koraterhességben NIPT teszt mellett vizsgálva rendkívül hasznos lehet.
A cisztás fibrózis és szűrése részletesen
Mi a cisztás fibrózis?
A cisztás fibrózis az egyik leggyakoribb örökölt genetikai betegség, amely változatos tünetekkel jelentkezik és több különböző szervet is érinthet. A betegség az egészen enyhe formától a súlyos tünetekig sokféle módon befolyásolhatja a szervezet működését. A cisztás fibrózis betegségtől a szervezet mirigyei a normálisnál sokkal sűrűbb váladékokat termelnek, életveszélyes tüdőfertőzéshez, emésztési problémákhoz, férfi meddőséghez vezetve.
Mik a cisztás fibrózis tünetei?
Cisztás fibrózisban a szervezet mirigyváladékainak összetétele megváltozik, mely jelenség az egész szervezet működésére kihatással van. A betegség általános tünete a verejték nagyon magas sótartalma, amely olykor sókristályok megjelenéséhez vezet a beteg bőrének felszínén. A tünetek súlyossága, valamint az érintett szervrendszerek köre egyénenként változhat és függ a betegséget okozó génhibáktól is. Súlyos esetekben a tüdő, a hasnyálmirigy, belek is érintettek, máskor a betegségnek pl. csak a hasnyálmirigyre vagy a reproduktív szervekre van hatása. Leggyakrabban a tüdő és az emésztőszervek érintettek a betegségben.
Mi történik a cisztás fibrózisos beteg tüdejében?
A tüdőben a betegség miatt eltérően működő sejtek az egészségesnél sűrűbb váladékot választanak ki, amely a tüdőhólyagocskák falát bevonva nehezíti a légzést, és köhögésre ingerel. A sűrű váladékban könnyen megtapadnak a kórokozók is, visszatérő tüdőfertőzésekhez vezetve.
Mi történik a cisztás fibrózisban szenvedő beteg emésztőrendszerében?
A hasnyálmirigy az emésztőrendszerünk fontos része, mely az elfogyasztott táplálék emésztését elősegítő enzimeket termel. Ez, az emésztőenzimek által lebontott táplálék szívódik fel később a bélben. Ha a betegség a hasnyálmirigyet is érinti, a mirigyen belüli kis csatornácskák a sűrű váladék miatt elzáródnak, aminek következtében az emésztőenzimek nem tudnak a bélbe ürülni, és a mirigyszövet is károsodik. Az emésztőenzimek hiányában a táplálék emésztetlenül halad át a tápcsatornán, a tápanyagok hasznosítatlanul ürülnek, a fejlődés a tápanyaghiány miatt elmarad. Idősebb korban a cisztás fibrózis következtében gyakran alakul ki inzulinfüggő cukorbetegség.
Hogyan befolyásolja a cisztás fibrózis a termékenységet?
A cisztás fibrózisban szenvedő férfiak 98%-a meddő, ez a tünet azonban nem feltétlenül párosul a cisztás fibrózis betegség egyéb tüneteivel. Bizonyos CFTR mutációk, melyek nem okoznak klasszikus CF tüneteket, fiúgyermekekben veleszületett ondóvezeték hiányhoz vezetnek. A kórképet a betegség angol nevének (congenital bilateral absence of vas deferens) rövidítéséből vett mozaikszóval CBAVD-nak is nevezzük. Az ondóvezeték hiánya miatt az érintett férfiak ejakulátuma nem tartalmaz spermiumokat (azoospermia). A férfi meddőség hátterében 1-2 százalékban ez a jelenség áll. Tekintettel arra, hogy az érintett férfiaknál nem a spermiumok termelődése, hanem azok ürülése szenved zavart, az érintett párok lombikprogramban sikeresen vehetnek részt.
A betegséggel érintett nőknél a sűrű méhnyakváladék miatt a teherbeesés az átlagosnál nehezebb.
Mikor gyanakodjunk cisztás fibrózisra?
A cisztás fibrózis tünetei változatosak lehetnek, és bármely életkorban jelentkezhetnek, de legtöbbször már kora gyermekkorban felfigyelhetünk rájuk.
Árulkodó jelek lehetnek:
- verejték nagyon magas sótartalma
- gyakori köhögés, sűrű felköhögött váladék
- gyakori tüdőgyulladás, ill. a tüdő megbetegedései
- alacsony testmagasság
- súlyvesztés jó étvágy ellenére
- emésztési panaszok
- újszülöttkori bélelzáródás
- férfi meddőség (azoospermia)
Mit kell tudni a cisztás fibrózis genetikai hátteréről?
A CFTR gén
A cisztás fibrózis kialakulása a CFTR gén mutációihoz köthető. Ez a gén egy olyan fehérjét kódol, amely a sejthártyába épülve klorid-ion csatornaként működik és a tüdő-, máj-, hasnyármirigy-, bél-, reproduktív traktus- és bőrsejtek membránjában felel a klorid-transzport folyamatokért. A CFTR gén igen hosszú és több mint 4000 különböző mutációt hordozhat. Ezekből a mutációkból kb. 1000 vezet betegség kialakulásához, a többi esetében a mutáció a fehérje működését nem, vagy nem jelentős mértékben befolyásolja, így tüneteket nem okoz. A mutációk gyakorisága populációnként változó, a leggyakoribb mutáció az F508del, melynek előfordulási gyakorisága, akárcsak a többi ismert CF mutációé földrajzi-etnikai varianciát mutat. Európán belül az F508del mutáció előfordulási gyakorisága délkelet-északnyugat irányú eltolódást mutat, Törökországban például a CF-es kromoszómáknak kb. 30% -án, míg Dániában 88%-án ez a mutáció mutatható ki.
Mit jelent az, hogy valaki cisztás fibrózis hordozó?
Minden ember két CFTR génnel rendelkezik, egyiket az egyik, másikat a másik szülőtől örököltük. Ha valakinek az egyik szülőtől kapott génje egészséges, a másik hibás, akkor az egyén cisztás fibrózis hordozó. Mivel a hordozó csak egy hibás génnel rendelkezik, nem lesznek tünetei, mert a másik szülőtől kapott, egészséges génje ellátja feladatát.
Hogyan öröklődik a ciszás fibrózis?
Az európai népességben jellemzően minden 25-ik ember hordoz valamilyen CF mutációt, míg pl. az ázsiai populációban csak minden 90-ik ember érintett. A cisztás fibrózis recesszív öröklődésű genetikai betegség, ami azt jelenti, hogy ahhoz, hogy valaki beteg legyen, mindkét szülőjétől hibás gént kell kapnia.
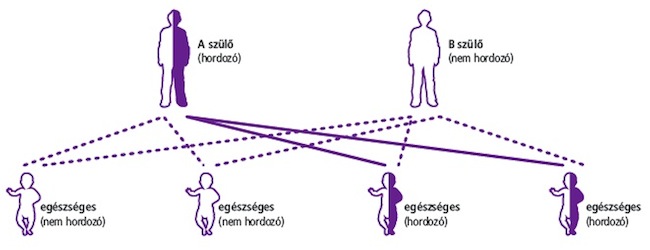
Ha ön cisztás fibrózis hordozó, de párja nem, gyermekei egészségesek lesznek, de 50%-os eséllyel hordozzák majd a cisztás fibrózist.
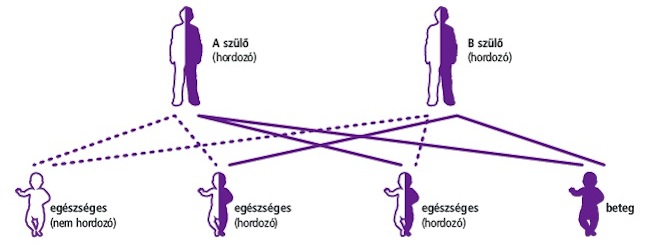
Ha ön is és párja is cisztás fibrózis hordozó, 25% az esélye annak, hogy a születendő gyermek Öntől is és partnerétől is a hibás gént örökli. Ilyenkor lehetőség van magzati korban végzett diagnosztikára, vagy lombikprogramban történő részvételre, és az embriók beültetés előtti vizsgálatára.
Bárki lehet cisztás fibrózis hordozó?
Igen. Európában ez a leggyakoribb öröklött genetikai betegség, minden 25. ember hordoz valamilyen cisztás fibrózis mutációt. Ha a családban előfordult már cisztás fibrózis betegség, akkor a hordozóság valószínűsége az átlagosnál nagyobb.
A cisztás fibrózis szűrése
Az, hogy Ön hordozza-e a cisztás fibrózist, az Istenhegyi Géndiagnosztikai Centrumban egy vérvizsgálatra épülő genetikai teszttel megállapítható. A cisztás fibrózis gyakorisága a Down-szindrómaénak mintegy negyede, de miközben a Down-szindróma szűrése lényegében kötelező eleme a terhesség alatti vizsgálatoknak, a cisztás fibrózis betegségről és szűrési lehetőségéről a családalapítás előtt álló szülők nagyobb része nem is tud.
Milyen előnyöm származik abból, ha részt veszek egy cisztás fibrózis szűrővizsgálaton?
Tekintve, hogy a cisztás fibrózis a leggyakoribb monogénes betegség, melynek minden 25. európai ember hordozója, mindenkinek 4% kockázata van arra, hogy hordoz egy cisztás fibrózis mutációt. Amennyiben két hordozó találkozik, minden terhességben 25% kockázatuk van arra, hogy a gyermeknél kialakuljon ez a súlyos betegség. Amennyiben legalább az egyik szülő részt vesz egy szűrővizsgálaton, mely negatív eredménnyel zárul, a kockázatuk jelentősen csökken. Ha Ön abba a 4%-ba tartozik, akik hordoznak valamilyen CF mutációt, fontos, hogy tudjon róla, hogy az átlagosnál nagyobb kockázattal rendelkezik, és párjával tudatosan tervezzék a gyermekvállalást, a partner hordozóságát is vizsgálva.
| Szülők eredménye | Kockázat arra, hogy a gyermek beteg lesz |
|---|---|
| egyik sem tesztelt | 1/2 500 |
| mindkettő tesztelt, egyik sem hordozó | 1/250 000 |
| 1 tesztelt nem hordozó, 1 nem tesztelt | 1/25 000 |
| mindkettő tesztelt, 1 hordozó, 1 nem hordozó | 1/1 000 |
| 1 tesztelt hordozó, 1 nem tesztelt | 1/100 |
| mindkettő tesztelt, hordozó | 1/4 |
Mit érdemes tudni a cisztás fibrózis szűrőtesztek hatékonyságáról általában? Miért válasszam a Géndiagnosztika tesztjét?
Fontos tudni, hogy a CFTR génnek több mint 4000 mutációja ismert, melyből kb. 1000 okoz cisztás fibrózist. A mutációk közül egyesek gyakoriak, míg mások lehetnek extrém ritkák (akár csak 1 családra jellemzőek). A szűrővizsgálatok azokra a mutációkra fókuszálnak, melyek populációs szinten minimum 1% körüli előfordulásúak. A cisztás fibrózis szűrés fontos kérdése, hogy a teszt milyen mutációkat vizsgál, ugyanis különböző populációkban különböző mutációk fordulnak elő gyakrabban. Így egy adott populációra kifejlesztett teszt érzékenysége egy más populáción jelentősen kisebb lehet. Vannak ugyanis olyan mutációk, melyek pl az amerikai népességben akár 5% körüli előfordulást is elérik, míg a magyar CF betegeknél még nem mutatták ki őket, és fordítva: vannak olyan közép-kelet európai eredetű mutációk, melyek előfordulási gyakorisága a magyar populációban 5% közeli, míg Amerikában elvétve fordulnak csak elő. Ezért fontos, hogy egy labor milyen teszttel dolgozik. Centrumunkban alkalmazott teszt detektációs rátája a magyar populációra nézve ~90%.
Mit tegyek, ha a családban már előfordult cisztás fibrózis?
Ha a családban már előfordult cisztás fibrózis, nagyon fontos, hogy gyermekvállalás előtt részt vegyen szűrővizsgálaton, hiszen az Ön rizikófaktora az átlagosnál magasabb. Amennyiben a vizsgálat Önnél is kimutatja a családban öröklődő mutáció jelenlétét, párjának a vizsgálatára is szükség lesz, hogy közös kockázatukat megállapítsuk. Amennyiben családi előzmény miatt jelentkezik a szűrésre, fontos, hogy kollégáink tudjanak a családi előzményről. A kérőlapon tüntesse fel, hogy milyen rokona (pl. nagynéni, unokatestvér gyermeke) érintett. Fontos továbbá, hogy az érintett családtagnál milyen mutációkat azonosítottak. Előfordulnak ugyanis olyan ritka mutációk, melyek csak egy-egy családban öröklődnek, így egy adott CFTR panelben nem feltétlenül szerepelnek. Ha ilyen mutáció van a családban, de mi nem tudunk róla, az félrevezető eredményt adhat, hiszen a panel nem fogja kimutatni a ritka mutációt, de a negatív eredmény ez esetben félrevezető lehet, hiszen azt a mutációs helyet, ahol a ritka mutáció elhelyezkedik, nem vizsgáltuk. Ha tudunk az előzményről, és tudjuk a mutáció típusát, kollégáink, ha kell, kiterjesztett szűrést végeznek a gén adott régiójára is a normál panelvizsgálaton felül. Így Ön biztos lehet abban, hogy az eredménye valóban informatív lesz, és megfelelő szakmai szempontok szerint történt a vizsgálat.
Mennyibe kerül a cisztás fibrózis szűrés?
A szűrővizsgálat aktuális árát árlistánkban találja. A szűrést centrumunk vérből vagy szájnyálkahártyából vett mintával végzi.
A cisztás fibrózis szűrés a Trisomy Super NIPT teszt részeként is kérhető, ebben az esetben a teszt módszertana és jellemzői némileg eltérnek. Erről részletes információ a kérőlapon olvasható, valamint a teszthez járó genetikai konzultáción kérhető.
Hogy értelmezzem a szűrés negatív eredményét?
Ha a szűrés eredménye negatív, akkor annak a valószínűsége, hogy cisztás fibrózis mutációt hordoz, jelentősen csökken. Ha a pár mindkét tagja negatív, annak kockázata, hogy a születendő gyermek cisztás fibrózis beteg lesz 1:2 500-ról 1:250 000-re csökken.
Hogy értelmezzem a szűrés pozitív eredményét?
Ha a teszt pozitív lesz, Ön hordozó. Pozitív eredmény esetén gyermekvállalás előtt feltétlenül szükséges genetikai tanácsadáson résztvenni. Ebben az esetben kiemelten fontos partnerénél is elvégezni a cisztás fibrózis tesztet, hiszen ettől az eredménytől jelentősen függ a kockázat mértéke. Amennyiben párjának negatív szűrési eredménye van, akkor közös kockázatuk CF beteg gyermek születésére 1/1 000-re (0,1%) csökken. Amennyiben azonban párja is hordozónak bizonyul, 25% kockázatuk van CF beteg gyermek születésére. Amennyiben Ön és párja is hordozó, a gyermekvállalással kapcsolatos kérdésekben kérjék szakembereink tanácsát.
Milyen mutációkat mutat ki a teszt?
A teszt az alábbi mutációk észlelésére validált:
1) F508del (c.1521_1523del, p. Phe508del)
2) I507del (c.1516_1518del, p.Ile507del)
3) G542X (c.1624G>T, p.Gly542*)
4) G551D (c.1652G>A, p.Gly551Asp)
5) R553X (c.1657C>T, p.Arg553*)
6) CFTRdele2,3 (21kb) (c.54-5940_273+10250del21080)
7) G85E (c.254G>A, p.Gly85Glu)
8) 1717-1G>A (c.1585-1G>A)
9) R117H (c.350G>A, p.Arg117His)
10) R117C (c.349C>T, p.Arg117Cys)
11) 621+1G>T (c.489+1G>T)
12) N1303K (c.3909C>G, p.Asn1303Lys)
13) W1282X (c.3846G>A, p.Trp1282*)
14) 1898+1G>A (c.1766+1G>A)
15) 2184insA (c.2052dup, p.Gln685Thrfs*4)
16) 2183AA>G (c.2051_2052delAAinsG, p.Lys684Serfs*38)
17) 3849+10kbC>T (c.3718-2477C>T)
18) Y1092X (c.3276C>A, c.3276C>G, p.Tyr1092*)
19) 3659delC (c.3528del, p.Lys1177Serfs*15)
20) R560T (c.1679G>C, p.Arg560Thr)
21) 2184delA (c.2052del, p.Lys684Asnfs*38)
22) R1066C (c.3196C>T, p.Arg1066Cys)
23) A455E (c.1364C>A, p.Ala455Glu)
24) D1152H (c.3454G>C, p.Asp1152His)
25) G622D (c.1865G>A, p.Gly622Asp)
26) 1677delTA (c.1545_1546del, p.Tyr515*)
27) 3272-26A>G (c.3140-26A>G)
28) 1078delT (c.948del, p.Phe316Leufs*12)
29) 711+1G>T (c.579+1G>T)
30) 3120+1G>A (c.2988+1G>A)
31) 2789+5G>A (c.2657+5G>A)
32) R1162X (c.3484C>T, p.Arg1162*)
33) R347H (c.1040G>A, p.Arg347His)
34) R347P (c.1040G>C, p.Arg347Pro)
35) R334W (c.1000C>T, p.Arg334Trp)
36) L206W (c.617T>G, p.Leu206Trp)
37) P5L (c.14C>T, p.Pro5Leu)
38) R1070Q (c.3209G>A, p.Arg1070Gln)
39) S945L (c.2834C>T, p.Ser945Leu)
40) 1154insTC (c.1019_1020dup, p.Phe342Hisfs*28)
41) 1548delG (c.1418del, p.Gly473Glufs*54)
42) 1811+1.6kbA>G (c.1680-886A>G)
43) 1898+5G>T (c.1766+5G>T)
44) 2789+2insA (c.2657+2_2657+3insA)
45) 3876delA (c.3744del, p.Lys1250Argfs*9)
46) 3905insT (c.3773dup, p.Leu1258Phefs*7)
47) 394delTT (c.262_263del, p.Leu88Ilefs*22)
48) A559T (c.1675G>A, p.Ala559Thr)
49) E60X (c.178G>T, p.Glu60*)
50) F311del (c.935_937del, c.933_935del, p.Phe311del)
51) G970D (c.2909G>A, p.Gly970Asp)
52) I618T (c.1853T>C, p.Ile618Thr)
53) M1101K (c.3302T>A, p.Met1101Lys)
54) P67L (c.200C>T, p.Pro67Leu)
55) Q493X (c.1477C>T, p.Gln493*)
56) Q890X (c.2668C>T, p.Gln890*)
57) R1070W (c.3208C>T, p.Arg1070Trp)
58) R1158X (c.3472C>T, p.Arg1158*)
59) R352Q (c.1055G>A, p.Arg352Gln)
60) R75X (c.223C>T, p.Arg75*)
61) S549N (c.1646G>A, p.Ser549Asn)
62) V456A (c.1367T>C, p.Val456Ala)
63) V520F (c.1558G>T, p.Val520Phe)
64) Y122X (c.366T>A, p.Tyr122*)
65) IVS8 polyT (c.1210-12T[5-9])
Gyakran ismételt kérdések
Ha a családban eddig még nem fordult elő a betegség, születhet cisztás fibrózis beteg gyermekem?
IGEN. A legtöbb család, ahová cisztás fibrózis beteg gyermek születik, korábban nem volt érintett.
Minden egyes gyermekvállaláskor újra el kell végeztetni a tesztet?
NEM. Ha Ön nem hordozó, nagyon kicsi a valószínűsége, hogy cisztás fibrózis beteg gyermeke születik. Ha Ön hordozó, és új partnere van, akkor az új partnernél érdemes a szűrővizsgálatot elvégezni.
Ha a szűrővizsgálat nem talál eltérést, lehetek még hordozó?
IGEN. A szűrés a hordozók közel 90%-át azonosítja, mivel csak a gyakoribb mutáció típusokat vizsgálja. Akinek a szűrővizsgálata negatív, kis valószínűséggel hordoz valamilyen ritka cisztás fibrózis mutációt. Ha mindkét szülő eredménye negatív a szűrővizsgálaton, kockázatuk 1:2 500-ról 1:250 000-re csökken.
Ha cisztás fibrózis hordozó vagyok, kifejlődik nálam a betegség?
NEM. Amennyiben Ön hordozó, a két cisztás fibrózisért felelős génjéből csak az egyik hibás, a másik viszont működőképes, így tünetei nem lesznek.
Mit tegyek, ha a családban cisztás fibrózis beteg vagy hordozó van?
Ebben az esetben az Ön rizikófaktora az átlagosnál nagyobb, így javasolt Önnél és partnerénél is elvégezni a szűrővizsgálatot. Pozitív eredmény esetén gyermekvállalás előtt feltétlenül szükséges genetikai tanácsadáson részt venni.
Genetika
A genetikai vizsgálatok ma már számos kérdésünkre adhatnak választ. Átadhatok-e tudtomon kívül betegséget a gyermekemnek? Állhat-e a genetika a háttérben, ha nem jön a baba? Kisgyermekemnél problémákat találtak, elképzelhető, hogy genetikai betegsége van? Örökölhettem rákra, vagy más betegségre való hajlamot? Várjuk genetikai tanácsadásunkon, hogy szakorvosaink javaslatai alapján késlekedés nélkül elindulhat a megfelelő úton!
Genetikai tanácsadás terhesség előtt »klinikai genetikussal: Dr. Tankó Lenke » | Dr. Varga Norbert » | 32 000 Ft |
Genetikai hordozóságszűrés gyermeket tervező pároknak (egy fő) » (1 fő)az ár tartalmazza a genetikai szaktanácsadást is, eredmény 1 hónap | 279 000 Ft |
Genetikai hordozóság szűrés gyermeket tervező pároknak (a pár mindkét tagjának egyidejű vizsgálata) »az ár tartalmazza a genetikai szaktanácsadást is, eredmény 1 hónap | 490 000 Ft |
Felnőtt spinális izomatrófia (SMA) hordozóságszűrő vizsgálat »genetikai tanácsadás, genetikai konzultáció szükséges | 49 000 Ft |
Preimplantációs Genetikai Diagnosztika (PGD)
genetikai tanácsadás szükséges | 60 000 Ft / ciklus + 30 000 Ft / embrió |
Preimplantációs Genetikai Diagnosztika (PGD)
genetikai tanácsadás szükséges | beállítási díj 650 000 Ft; 130 000 Ft / ciklus + 75 000 Ft / embrió |
Kariotipizálás (citogenetikai kromoszóma vizsgálat) »vérvétel minden hétköznap 9:00-14:30 óra között, előzetes időpontfoglalás szükséges, eredmény 6-8 hét | 45 000 Ft |
Y-kromoszóma mikrodeléció (AZF) vizsgálat | 49 000 Ft |
Genetikai konzultáció és tanácsadás, genetikai hordozóságszűrés gyermeket tervező pároknak, menedzsergénszűrés, a laktóz- és gluténérzékenység genetikai vizsgálata, trombózishajlam-vizsgálat és HLA-B27 meghatározás reumás betegségek, krónikus ízületi gyulladás, ízületi fájdalom vagy merevség jelenléte, illetve különféle autoimmun betegségek gyanúja esetén.
Genetikai tanácsadás »klinikai genetikussal: Dr. Tankó Lenke » | Dr. Varga Norbert » | 32 000 Ft |
Genetikai hordozóságszűrés gyermeket tervező pároknak (egy fő) » (1 fő)az ár tartalmazza a genetikai szaktanácsadást is, eredmény 1 hónap | 279 000 Ft |
Genetikai hordozóság szűrés gyermeket tervező pároknak (a pár mindkét tagjának egyidejű vizsgálata) »az ár tartalmazza a genetikai szaktanácsadást is, eredmény 1 hónap | 490 000 Ft |
Felnőtt spinális izomatrófia (SMA) hordozóságszűrő vizsgálat »genetikai tanácsadás, genetikai konzultáció szükséges | 49 000 Ft |
Újszülöttkori Spinális izomatrófia (SMA) betegség szűrővizsgálata »genetikai tanácsadás, genetikai konzultáció szükséges | 38 000 Ft |
Menedzsergén-szűrés – Apo E genotípus meghatározás »
18 év alatt orvosi beutaló szükséges | 30 000 Ft |
Gluténérzékenység genetikai hajlam vizsgálat »18 év alatt orvosi beutaló szükséges | 33 000 Ft |
Laktózérzékenység vizsgálat »18 év alatt orvosi beutaló szükséges, csecsemőknél fájdalommentes szájnyálkahártya törlet | 16 500 Ft |
Trombózishajlam vizsgálat »V. faktor Leiden, MTHFR C677T, MTHFR A1298C, II. faktor Prothrombin gén mutációk 18 év alatt orvosi beutaló szükségesVérvétel minden hétköznap 9:00-14:30 óra között, előzetes időpontfoglalás szükséges. | 30 000 Ft |
V. faktor Leiden mutáció | 12 500 Ft |
II. faktor (prothrombin) G20210A mutáció | 12 500 Ft |
MTHFR C677T mutáció | 12 500 Ft |
MTHFR A1298C mutáció | 12 500 Ft |
HLA-B27 antigén meghatározás »18 év alatt orvosi beutaló szükséges | 29 000 Ft |
Genetikai tanácsadás beteg gyermeknél és genetikai konzultáció, génpanel vizsgálat feltételezett genetikai betegség esetén. Centrumunkban többféle ritka betegség (például Fragilis X-szindróma, Duchenne/Becker izomdisztrófia, spinális izomatrófia) genetikai vizsgálata is biztosított.
Genetikai tanácsadás beteg gyermek esetén »klinikai genetikussal: Dr. Tankó Lenke » | Dr. Varga Norbert » | 32 000 Ft |
Genetikai konzultációhumángenetikussal: Dr Karcagi Veronika » | 25 000 Ft |
Öröklődő genetikai rendellenességek vizsgálata »genetikai tanácsadás, genetikai konzultáció szükséges | egyedi ár |
Spinális izomatrófia (SMA) genetikai vizsgálata »genetikai tanácsadás, genetikai konzultáció szükséges | 49 000 Ft |
Cisztás fibrózis szűrés »genetikai tanácsadás szükséges, eredmény 7-10 munkanap | 85 000 Ft |
Fragilis X-szindróma genetikai vizsgálata »genetikai tanácsadás szükséges, eredmény 8 hét | 105 000 Ft |
Genetikai tanácsadás beteg gyermeknél és genetikai konzultáció, génpanel vizsgálat feltételezett genetikai betegség esetén. Centrumunkban többféle neurológiai betegség (öröklődő perifériás neuropátiák, Leber-féle öröklődő optikus neuropátia, öröklődő végtagi izomdisztrófiák) genetikai vizsgálata is biztosított.
Genetikai tanácsadás »klinikai genetikussal: Dr. Tankó Lenke » | Dr. Varga Norbert » | 32 000 Ft |
Genetikai konzultációhumángenetikussal: Dr Karcagi Veronika » | 25 000 Ft |
Gyermekneurológiai szakorvosi kontroll vizsgálat » | 36 000 Ft |
Gyermekneurológiai szakorvosi vizsgálatDr.Herczegfalvi Ágnes osztályvezető egyetemi docens rendelésénAz ár az esetlegesen javasolt laborvizsgálatok árát nem tartalmazza | 25 000 Ft |
Génpanel vizsgálat – hasonló klinikai tüneteket okozó gének egyidejű analízisére »genetikai tanácsadás, genetikai konzultáció szükséges | egyedi ár |
Spinális izomatrófia (SMA) genetikai vizsgálata »genetikai tanácsadás, genetikai konzultáció szükséges | 49 000 Ft |
Újszülöttkori Spinális izomatrófia (SMA) betegség szűrővizsgálata »genetikai tanácsadás, genetikai konzultáció szükséges | |
Duchenne/Becker-féle izomdisztrófia (DMD) genetikai vizsgálata »genetikai tanácsadás, genetikai konzultáció szükséges | 165 000 Ft |
Fragilis X-szindróma genetikai vizsgálata »genetikai tanácsadás szükséges, eredmény 8 hét | 105 000 Ft |
Öröklődő perifériás neuropátiák genetikai vizsgálata »genetikai tanácsadás, konzultáció szükséges | egyedi ár |
Öröklődő végtagövi izomdisztrófiák genetikai vizsgálata »genetikai tanácsadás, konzultáció szükséges | egyedi ár |
Leber-féle öröklődő optikus neuropátia genetikai vizsgálata »genetikai tanácsadás, konzultáció szükséges | egyedi ár |
Miotóniás disztrófiák - Dystrophia myotonica (DM1 és DM2) genetikai vizsgálata »genetikai tanácsadás, konzultáció szükséges | egyedi ár |
Facioscapulohumeralis izomdisztrófia (FSHD) genetikai vizsgálata »genetikai tanácsadás, konzultáció szükséges | egyedi ár |
Az Istenhegyi Géndiagnosztikai Centrum célja, hogy az elérhető legátfogóbb genetikai szűrővizsgálatot nyújtsa az örökletes rákhajlamosító mutációk felderítésére. 134 gént vizsgáló génpanelünkkel a legtöbb jelenleg ismert ilyen gén vizsgálható. Öröklődő emlő- és petefészekrákhajlam szűrés, öröklődő vastagbélrákhajlam szűrés.
Genetikai tanácsadás »klinikai genetikussal: Dr. Tankó Lenke » | Dr. Varga Norbert » | 32 000 Ft |
Örökletes rákhajlam komplex csomag – emlőrák és petefészekrák »Részletes előzetes onkológiai és genetikai szaktanácsadás, komplex molekuláris genetikai vizsgálatés utólagos onkológia és genetikai szaktanácsadás | 360 000 Ft |
Öröklődő mell-és petefészekrák szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő prosztatarák szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő gyomorrák szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő veserák szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő multiplex endokrin neoplázia szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő pajzsmirigyrák szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő mellékpajzsmirigyrák szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Neurofibromatózis (NF1, NF2) szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő retinoblasztóma szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Pheokromocytoma szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő familiáris paraganglioma szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő melanoma szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő chondrosarcoma szűrése »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő vastagbélrák szűrés »az ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő hasnyálmirigyrák szűrésaz ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Öröklődő méhnyálkahártyarák szűrésaz ár tartalmazza a genetikai szaktanácsadást is | 290 000 Ft |
Kardiológiai genetikai vizsgálatunk során Ön átfogó képet kaphat arról, hogy érintett lehet-e örökletesen előforduló szívbetegségben. Vizsgálatunk kiemelt területe a szívizom-elfajulások, más néven kardiomiopátiák genetikai kockázatának előrejelzése.
Genetikai tanácsadás »klinikai genetikussal: Dr. Tankó Lenke » | Dr. Varga Norbert » | 32 000 Ft |
Genetikai hajlamszűréssel kombinált kardiológiai szűrőcsomag » | 111 000 Ft |
Menedzsergén-szűrés – Apo E genotípus meghatározás »
18 év alatt orvosi beutaló szükséges | 30 000 Ft |
Trombózishajlam vizsgálat »V. faktor Leiden, MTHFR C677T, MTHFR A1298C, II. faktor Prothrombin gén mutációk 18 év alatt orvosi beutaló szükségesVérvétel minden hétköznap 9:00-14:30 óra között, előzetes időpontfoglalás szükséges. | 30 000 Ft |
V. faktor Leiden mutáció | 12 500 Ft |
II. faktor (prothrombin) G20210A mutáció | 12 500 Ft |
MTHFR C677T mutáció | 12 500 Ft |
MTHFR A1298C mutáció | 12 500 Ft |